Workflow¶
The workflow of the pipeline is shown below:
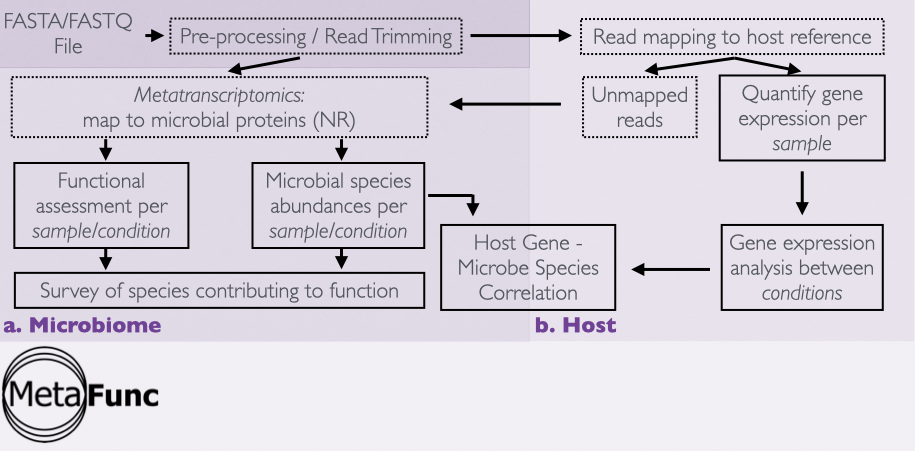
Fig. 1 MetaFunc workflow¶
The pipeline is executed in order below starting with sequencing reads in fasta- or fastq-format stemming from a transcriptomic or genomic experiment. It is also indicated if a step is optional - there is an option in the config file not to go through with the step.
Initial processing of inputs (OPTIONAL)
Note
The entire pre-processing of inputs is optional. A user can start at any point in the 3 steps below. For step 3, one can either use unmapped reads from step 2 or directly input reads to the microbiome analysis if the reads are already of satisfactory quality and do not require adapter and/or host read removal.
Use fastp for adapter trimming and quality control
Use trimmed reads in STAR to map to a host genome
Use (unmapped) reads as input to Kaiju
Host Analysis (OPTIONAL)
Note
Reads are from step 2 of Initial processing of inputs. DGEA and GSEA are only performed if comparisons between groups are indicated. To perform a GSEA an additional gene set file in GMT-format is needed (a human Gene Ontology file is included as an example).
Use featureCounts of the subread package to quantify gene counts
Use gene count information to perform DGEA using edgeR. (OPTIONAL)
Use information obtained from DGEA to perform GSEA using clusterProfiler. (OPTIONAL)
Microbiome Analysis
Note
Reads are from step 1 or 3 of Initial processing of inputs. This part of the workflow is mandatory but analyses based on group/condition information are optional.
Taxonomy and protein matches per read are identified using Kaiju.
Taxonomy ID counts per sample are summarized into a table per sample.
Table with Taxonomy ID counts per group is generated (OPTIONAL).
Use TaxID and group information to get differentially abundant species using edgeR. (OPTIONAL)
Use protein information to get gene ontology (GO) annotations for each sample.
Tables with GOs per group/condition are generated (OPTIONAL).
Host - Microbiome Relationship (OPTIONAL)
Note
Input to this step are the top DGEs (specifiable in (4h) of the Adjust config.yaml) from step 2 of Host Analysis and top DA species (specifiable in (4h) of the Adjust config.yaml) from step 2b of Microbiome Analysis.
Spearman correlation analysis between DEGs of the host gene analysis and DA species of the microbiome taxonomy analysis is performed
Results are summarized in a matrix and clustered using clustergrammer.